Use case test
One of the possible uses of NGSphy might be the optimization of depth of coverage for a given purpose. In this case we designed a small experiment to visualize the potential trade-off between NGS coverage and SNP discovery. In this case we used NGSphy to simulate a single sequence alignment from a given gene-tree (Figure 1) and from it we generated 100 NGS datasets at different depths of coverage.
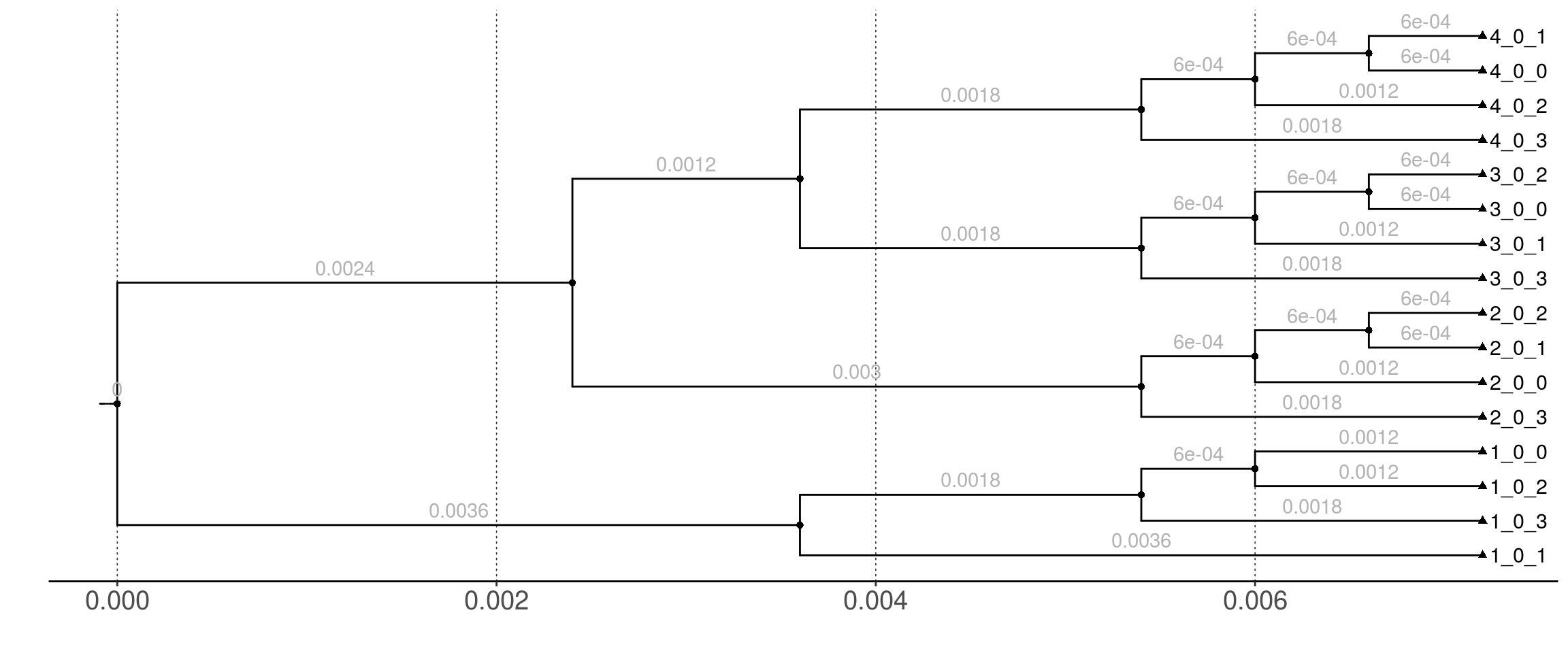
Figure 1: Gene-tree used for the use case simulation. It represents four species with two individuals per species. Numbers above the branches represent branch lengths, in expected number of substitutions.
The sequence alignments were simulated under a JC69 substitution model (Jukes and Cantor, 1969), with equal base frequencies and a length of 10.000 bp. The Illumina runs generated 150 bp paired-end reads for all individuals at a coverage of 2X, 10X, 50X, 100X and 200X (100 replicates for each level). The detailed settings are shown in Table 1.
Table 1: NGSphy settings parameters. varies for 2, 10, 20, 100 and 200x.
| Block | Option | Value |
|---|---|---|
| general | path | ./output/<coverage> |
| output_folder_path | NGSphy_test2_<coverage> |
|
| ploidy | 1 | |
| data | inputmode | 1 |
| gene_tree_file | files/supp.test2.tree |
|
| indelible_control_file | files/control.supp.test2.txt |
|
| coverage | experiment | <coverage> |
| ngs-reads-art | fcov | true |
| l | 150 | |
| m | 250 | |
| p | true | |
| q | true | |
| s | 50 | |
| ss | HS25 | |
| execution | environment | bash |
| runART | on | |
| threads | 2 |
Mapping was carried out using the MEM algorithm of BWA Version 0.7.7-r441 (Li and Durbin, 2009),
against a randomly chosen reference (sequence 1_0_2 in all cases). Following a
standardized best-practices pipeline (Van der Auwera and Carneiro, 2013) mapped reads
from all replicates were independently processed, performing local realignment around
indels and removing PCR duplicates. Variant calling was made with GATK (Mckenna et al., 2010),
using the single-sample variant-calling joint-genotyping framework using the HaplotypeCaller
and GenotypeVCF modules. SNP calls from each replicate were compared to the true variant
sites, showing that SNP recovery increased very rapidly until 10X, when almost all true
variants were called (Figure 2).

Figure 2: Called variants and true positives (mean and Q1/Q3) at different depths of coverage. The true number of SNPs is 386. At 100x and 200x only 1 site (average) is not recovered.
The script use to carry out this test can be found under ngsphy/manuscript/supp.material/scripts/supp.test2.sh.
To run this test you need the following files:
- NGSphy settings file (2x):
ngsphy/data/settings/ngsphy.settings.supp.test2.2x.txt - NGSphy settings file (10x):
ngsphy/data/settings/ngsphy.settings.supp.test2.10x.txt - NGSphy settings file (20x):
ngsphy/data/settings/ngsphy.settings.supp.test2.20x.txt - NGSphy settings file (100x):
ngsphy/data/settings/ngsphy.settings.supp.test2.100x.txt - NGSphy settings file (200x):
ngsphy/data/settings/ngsphy.settings.supp.test2.200x.txt - NGSphy settings file (200x - read counts):
ngsphy/data/settings/ngsphy.settings.supp.test2.200x.rc.txt - INDELible control file:
ngsphy/data/indelible/control.supp.test2.txt - Tree file:
ngsphy/data/trees/supp.test2.tree - Referene allele file for read counts:
ngsphy/data/reference_alleles/my_reference_allele_file.test2.txt
To execute this script it is required to have installed:
- Following the script file, first we have to state:
- Where the NGSphy repository is located
- Where will the output be written
- The name of the output folder
- A random seed number
- Paths for GATK and PICARD jar packages
- Coverage levels
- Organize the data to run the simulation and the analysis, copying the setting files, reference sequence and the reference allele file into the analysis folder.
- Extract the true variants from the dataset, and we do that using NGSphy with the NGS read counts
- Select the sequence with the label
1_0_0, and put it into a separate file, this will be the reference sequence. - Run 100 replicates of NGSphy with the different coverage levels, each:
- Index the reference with
bwa,samtoolsandPicard: - Map all the datasets to the selected reference:
- Sort and covert into BAM file
- Mark duplicates
- Realignm around indels
- Single sample variant calling with each dataset, using HaplotypeCaller mode GCVF
- Call joint genotypes with GenotypeGVCFs
- Count discovered variants
- Compare discovered variatns with true variants
- Jukes, T.H. and Cantor, C.R. (1969) Evolution of Protein Molecules. In, Mammalian Protein Metabolism., pp. 21–132.
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760.
- Mckenna, A. et al. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20, 1297–1303.
- Van der Auwera, G.A. and Carneiro,M.O. (2013) From FastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Current protocols in Bioinformatics.