Dynamo workflow
Please go to our readthedocs webpage for more updated information: https://dynamo-release.readthedocs.io/en/latest/ten_minutes_to_dynamo.html
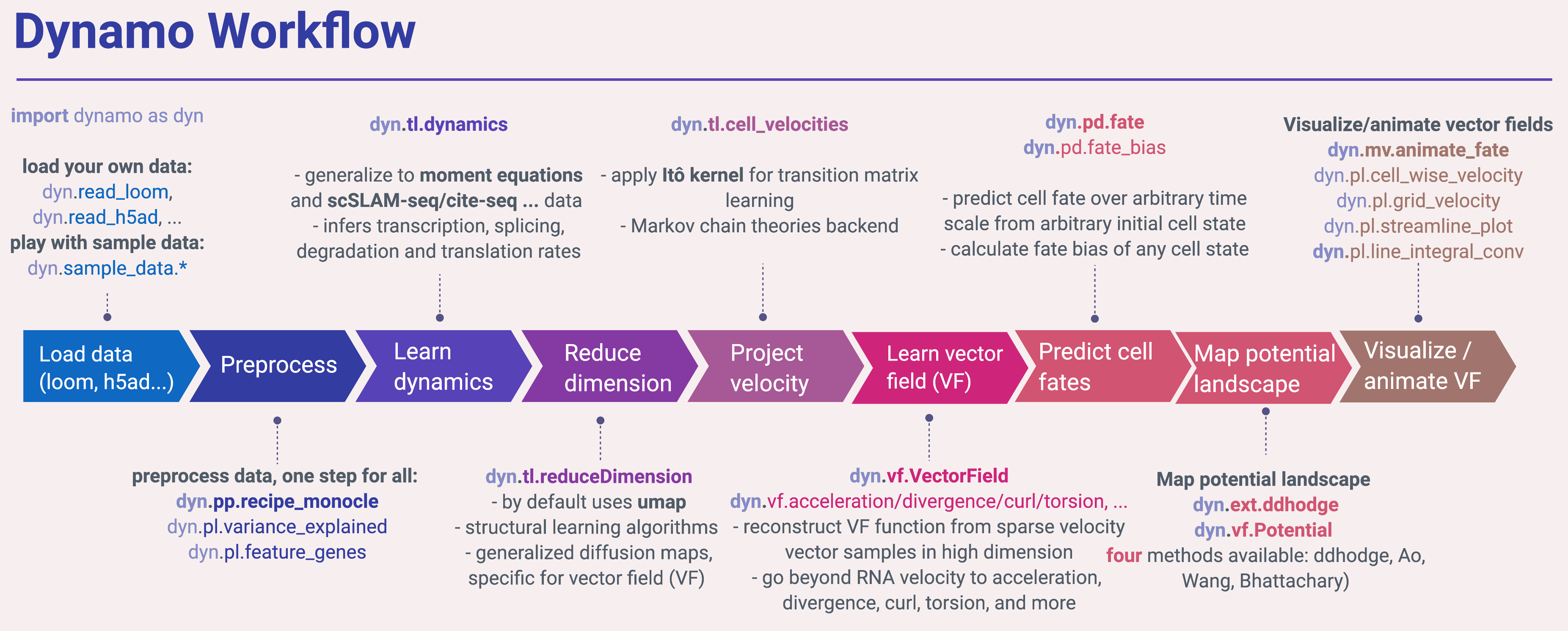
In the following, you can see python snippets that highlight each of the main steps in dynamo.
Surely the first step in applying dynamo is to load some datasets of interests. Dynamo uses AnnData as its container so you can load loom, h5 or h5ad, etc. files that generated from Kallisto + bustool or kb-python, loompy, velocyto, scanpy or even 10x cellranger (because dynamo can be reduced as a regular scRNA-seq analysis toolkit) via read_loom, read_h5, read_h5ad. Dynamo also provides a function (read_NASC_seq) to load output files
from the NASC-seq pipeline that applied to single cell based SLAM-seq/NASC-seq/Sci-fate/scNT-seq experiments. If you want to learn
protein velocity, you should then attach the protein abundance data to the .obsm attribute of the AnnData object via the protein key.
# first step: Load data
import dynamo as dyn
# set matplotlib's rcParams. this setting tries to emulate ggplot style.
dyn.configuration.set_figure_params('dynamo', background='black') # when background set to black, it produces cool figure that is good for presentation. set background='white' if you are producing figures for publication.
# you can read your own data via read_loom, read_h5ad or read_h5
adata = dyn.read_loom(filename, **param) # (or use read_h5ad, read_NASC_seq to integrate with scanpy work flow, load result generated from NASC-seq pipeline, etc.)
# here let us play with Dentate Gyrus example dataset (27, 998 genes and 18, 213 cells)
adata = dyn.sample_data.DentateGyrus() # there are many sample datasets available. You can check our tutorials for other available datasets: https://github.com/aristoteleo/dynamo-tutorials Next you may want to first check the fraction of the spliced, unspliced or new (metabolic labelled) or total mRNAs in your data by show_fraction. Then you are ready to run the recipe_monocle function that performs preprocessing of the data in a single shot. recipe_monocle uses similar strategy from Monocle 3 to normalize all datasets in different layers (the spliced and unspliced or new (metabolic labelled) and total mRNAs or others), followed by feature selection and PCA dimension reduction. You can check the mean-dispersion plot to see where do the automatically selected feature genes locate and also the variance explained plot to take a glance at the variance explained by top principal components. We also provide another preprocessing recipe (via recipe_velocyto) that is based on the seminal velocyto work. recipe_velocyto can be used to perform processing in a way that is (nearly) identical to the original velocyto package.
# second step: preprocess
dyn.pl.show_fraction(adata)
dyn.pp.recipe_monocle(adata)
dyn.pl.feature_genes(adata)
dyn.pl.variance_explained(adata)Next you will want to learn the velocity values for all genes that pass some filters (default it is all the selected feature genes) across cells. The dyn.tl.dynamics does all the hard work for you. It automatically checks the data you have and determine the experimental type automatically, either the conventional scRNA-seq, kinetics, degradation or one-shot single-cell metabolic labelling experiment or the cite-seq or reap-seq co-assay, etc. Then it learns the velocity for each feature gene using either the original deterministic model based on a steady-state assumption from the seminal RNA velocity work or a few new methods (see part of the derivations related to those models), that are based on moment equations which basically considers both mean and uncentered variance of gene expression. The second model requires the calculation of the first and second moment of the expression data that is based on a nearest neighbours graph, constructed in the reduced PCA space from the spliced or total mRNA expression. To minimize the distortion of expression dynamics, we can also use the mutual nearest neighbours graph that is shared between both the spliced or total mRNA PCA space and the unspliced or new mRNA one. Note that the first moment can be also treated as one local averaging (smoothing) of the original data, and thus it is used for estimating velocity vector even applying the first deterministic model for the purpose of denoising.
dyn.tl.dynamics not only learns degradation rates and the velocity vectors of spliced RNA as velocyto but also other possible kinetics parameters and velocity vectors according to your data automatically. For example, if you have metabolic labelling data (identified by the new and total or uu, ul, su and sl layers, etc.), dyn.tl.dynamics will learn the transcription, splicing and degradation rates for each gene and velocity vector for unspliced, spliced or new or old RNAs, etc.
# third step: learning dynamics
dyn.tl.dynamics(adata)You would like to also use different dimension reduction approaches to reduce your scRNA-seq into low dimensional embedding. By default, we use the popular umap algorithm but also supports other methods, for example, the novel trimap dimension reduction method. Trimap uses triplet constraints to form a low-dimensional embedding of a set of points which may be potentially more scalable and better at preserving the global structure of the data than UMAP. Note that we also generalized diffusion map so we can perform dimension reduction via diffusion while also considering drift. A class of structural learning-based dimension reduction methods developed by us (for example, probabilistic graph learning or PSL) will also be supported shortly.
# fourth step: reduce dimension
dyn.tl.reduceDimension(adata)You love to see the velocity vector on low dimensional embedding. To get there, you want to first apply our improved transition matrix reconstruction method. In contrast to the "correlation kernel" from the excellent velocyto and scVelo packages, dynamo is powered by the Itô kernel that not only considers the correlation between the vector from any cell to its nearest neighbours and its velocity vector but also the corresponding distances. We expect this new kernel will enable us to visualize more intricate vector flow or steady states in low dimension. We also expect it will improve the calculation of the stationary distribution or source states of sampled cells.
# fifth step: project velocity
dyn.tl.cell_velocities(adata)At this stage, you may already cannot wait to see how does the velocity vector looks like in low dimension space. Similar to velocyto and scvelo, we provide three plotting utilities that visualize the cell-wise velocity vector, velocity vector on a grid or the streamline plot that integrates along grid velocity vectors via fourth-order Runger-Kutta algorithm. In addition, we also provide a fourth method to visualize the structure of vector field, line integral convolution method or LIC (Cabral and Leedom 1993). LIC is an intuitive way to visualize vector field topography, which works by adding random black-and-white paint sources on the vector field and letting the flowing particle on the vector field picking up some texture to ensure the same streamline having similar intensity.
# sixth step: visualize vector field
color_list = ['Rgs20', 'Eya1', 'ClusterName'] # can include either gene, columns in the adata.obs attribute
dyn.pl.cell_wise_velocity(adata, color=gene_list, basis=basis, ncols=3)
# if you want to see cell wise quiver more clearly, try increase the figure size and quiver size and length, via figsize, quiver_size, quiver_length.
dyn.pl.grid_velocity(adata, color=gene_list, basis=basis, ncols=3, figsize=[8, 8])
dyn.pl.streamline_plot(adata, color=gene_list, ncols=3, figsize=[8, 8], density=3) It is often important to check velocity of a few marker genes and compare it with the gene expression to make sure whether the learnt velocity makes sense. We provide the dyn.pl.phase_portrait function can show the famous spliced-unspliced (or total-new, etc.) phase portrait, along with the velocity and gene expression for genes of interests.
genes = adata.var.index[adata.var.use_for_velocity][:3]
dyn.pl.phase_portraits(adata, genes=genes, ncols=3, figsize=(3, 3))
Note that you may find your favourite genes doesn't pass the gene filtering and thus don't have velocity in dynamo. To get the velocity for those genes anyway, you can actually run the following to calculate velocity for all genes, albeit it may take significantly more time and those velocity genes won't be used for downstream 2D velocity vector field embedding.
dyn.tl.dynamics(adata, filter_gene_mode='no')
Obviously we don't want to stop here. Let us move to the really exciting part of dynamo in the next section to learn the velocity vector in the full transcriptomic space and to map the potential landscape.
Dynamo aspires to learn a vector field function in the transcriptomic space. With the learnt vector field, you can then predict the cell fate in high dimension over arbitrary time scale from arbitrary initial cell states. You can experiment it via the dyn.tl.VectorField or dyn.tl.fate function.
# seventh step: learn vector field
dyn.tl.VectorField(adata)
dyn.pl.grid_velocity(adata, color=gene_list, ncols=3, method='SparseVFC')
dyn.pl.grid_velocity(adata, color=gene_list, ncols=3, method='SparseVFC')
dyn.pl.stremline_plot(adata, color=gene_list, ncols=3, method='SparseVFC')
dyn.pl.line_integral_conv(adata)Since we learn the vector function of the data, we can then characterize the topology of the full vector field space. For example, we are able to identify the fixed points (attractor/saddles, etc.) which may corresponds to terminal cell types or progenitors, nullcline, separatrices of a recovered dynamic system, which may formally define the dynamical behaviour or the boundary of cell types in gene expression space. To better visualize the topography of the learnt vector field, we provide the dyn.pl.topography function to visualize the structure of the 2D vector fields.
dyn.tl.topography(adata, dims=[0, 1])
dyn.pl.topography(adata)
Potential landscape is an intuitive concept that is widely used in various disciplines. It provides a global description of cell state stability. Once we learnt vector field, dynamo allows you to map the potential landscape (which is mathematically related to our favourite Waddington landscape model) and the least action paths that convert from any cell types (states) to any other cell types (states) with the highest probability. You can experiment it via the dyn.tl.Potential and
the dyn.tl.action function.
# eigth step: map potential landscape
dyn.tl.action(adata)
dyn.pl.Potential(adata)Note that this is our first beta version of Dynamo (as of Feb 25th, 2020). Dynamo is still under active development. Stable version of Dynamo will be released and submitted to PyPi when it is ready. Until then, please use Dynamo with caution and let us know if you confront any issues when using Dyanmo. We welcome any bugs reports (via GitHub issue reporter) and especially code contribution (via GitHub pull requests) of Dynamo from users to make it an accessible, useful and extendable tool. For discussion about different usage cases, comments or suggestions related to our manuscript and questions regarding the underlying mathematical formulation of dynamo, we provided a google group goolge group. Dynamo developers can be reached by xqiu.sc@gmail.com.
Dynamo requires python > 3.6. To install the newest version of dynamo, you can git clone our repo and then use:
git clone https://github.com/aristoteleo/dynamo-release.git
pip install dynamo-release/ --user Note that --user flag is used to install the package to your home directory instead, in cases you don't have the root privilege.
Alternatively, You can install Dynamo when you are in the dyname-release folder
git clone https://github.com/aristoteleo/dynamo-release.git
cd dynamo-release/
python setup.py install --user from source, using the following script:
pip install git+https://github.com:aristoteleo/dynamo-releaseIn order to ensure dynamo run properly, you need to ensure your python environment satisfies dynamo's dependencies. Especially, numpy and pands are to be numpy>=1.18.1, pandas>=0.25.1.
Xiaojie Qiu, Yan Zhang, Dian Yang, Shayan Hosseinzadeh, Li Wang, Ruoshi Yuan, Song Xu, Yian Ma, Joseph Replogle, Spyros Darmanis, Jianhua Xing, Jonathan S Weissman (2019): Mapping Vector Field of Single Cells. BioRxiv
biorxiv link: https://www.biorxiv.org/content/10.1101/696724v1
For the vector field reconstruction and potential landscape mapping, please refer to our preprint. We also released the derivation of the matrix form of the moment generating functions for parameter estimation in full_derivation.pdf file in the dynamo-notebook GitHub repo.
The dynamo-notebook repo also provides (outdated) tutorials on how to use dynamo for reconstructing vector field, calculating least action path and potential of cell states. The dynamo-tutorials repo provides various tutorials that are based on latest dynamo package.