- Clustering on Network of Samples
- Installation
- Installing Conos as Docker Container
- Usage example
- Reference
-
What is Conos? It's a package to wire together large collections of single-cell RNA-seq datasets. It focuses on uniform mapping of homologous cell types across heterogeneous sample collections. For instance, a collection of dozens of peripheral blood samples from cancer patients, combined with dozens of controls. And perhaps also including samples of a related tissue, such as lymph nodes.
-
How does it work?
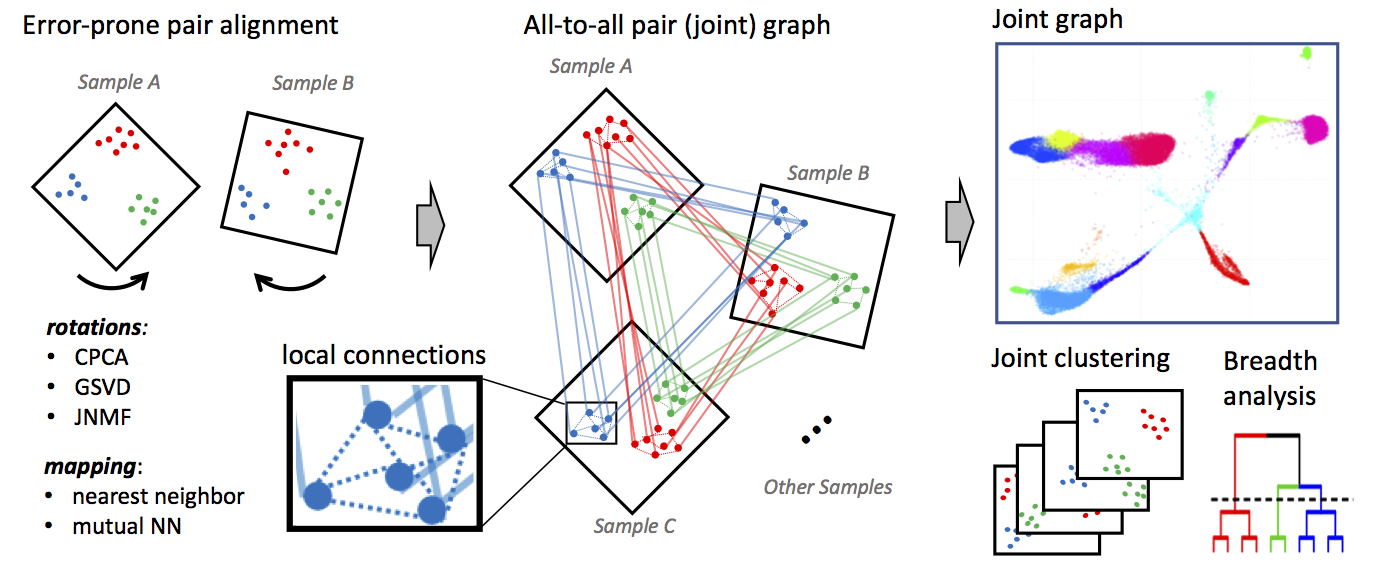
Conos applies one of many error-prone methods to align each pair of samples in a collection, establishing weighted inter-sample cell-to-cell links, creating a global joint graph. Cells of the same type will tend to map to each other across many such pair-wise comparisons, forming cliques, that can be recognized as clusters (graph communities).
-
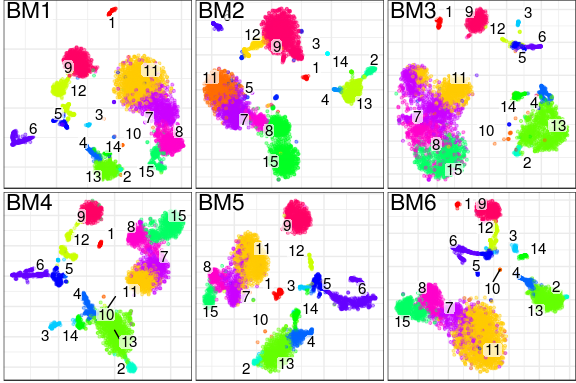
What does it produce? In essense, Conos will take a large, potentially heterogeneous panel of samples and will produce clustering grouping similar cell subpopulations together in a way that will be robust to inter-sample variation:
-
What are the advantages over existing alignment methods? Conos is robust to heterogeneity of samples within collection, as well as noise. The ability to resolve finer subpopulation structure improves as the size of the panel increases.
-
What do I need to run it? Conos is an R package. Currently, it supports pre-processing (filtering, normalization, etc.) of the individual datasets using pagoda2 or Seurat.
Native installations have been tested in Linux. Normal installation should take <10min.
Please make sure devtools package is installed (use install.packages("devtools") to install it if needed).
Then install pagoda2 (or Seurat), then install conos:
devtools::install_github("hms-dbmi/conos")The dependencies are inherited from pagoda2:
Install system dependencies, example here provided for Ubuntu
sudo apt-get update
sudo apt-get -y install libcurl4-openssl-dev libssl-devyum install openssl-devel libcurl-develIt is possible to install pagoda2 and Conos on OS X, however some users have reported issues with OpenMP configuration. For instructions see pagoda2 readme.
If your system configuration is making it difficult to install Conos natively, an alternative way to get Conos running is through a docker container.
Note: on OS X, Docker Machine has Memory and CPU limit. To control it, please check instructions either for CLI or for Docker Desktop.
The docker distribution has the latest version and also includes the Pagoda2 package. To start a docker container, first install docker on your platform and then start the pagoda container with the following command in the shell:
docker run -p 8787:8787 -e PASSWORD=pass docker.io/vpetukhov/conos:latest
The first time you run the command it will download several images so make sure that you have fast internet access setup. You can then point your browser to http://localhost:8787/ to get an Rstudio environment with pagoda2 and conos installed (log in using credentials rstudio / pass). Explore the docker --mount option to allow access of the docker image to your local files.
Note: if you already downloaded the docker image and want to update it, please run
docker pull vpetukhov/conos:latest
If you want to build image by your own, download the Dockerfile (available in this repo under /dockers) and run to following command to build it:
docker build -t conos .
This will create a "conos" docker image on your system (be patient, as the build takes ~30-50min or so). You can then run it using the following command:
docker run -d -p 8787:8787 -e PASSWORD=pass --name conos -it conos
Please see Conos tutorial for detailed usage. The overall runtime of the tutorial should be ~5 minutes.
Additional examples: forcing better alignment, integrating RNA-seq and ATAC-seq.
Given a list of individual processed samples (pl), Conos processing can be as simple as this:
# construct conos object, where pl is a list of pagoda2 objects
con <- Conos$new(pl)
# build graph
con$buildGraph()
# find communities
con$findCommunities()
# plot joint graph
con$plotGraph()
# plot panel with joint clustering results
con$plotPanel()For integration with ScanPy you need to save Conos files on disk from R session, than upload these files from Python. See the following tutorials:
If you find this pipeline useful for your research, please consider citing the paper:
Barkas N., Petukhov V., Nikolaeva D., Lozinsky Y., Demharter S., Khodosevich K. & Kharchenko P.V. Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nat. Methods, (2019). doi:10.1038/s41592-019-0466-z