-
Notifications
You must be signed in to change notification settings - Fork 0
/
README.Rmd
111 lines (82 loc) · 3.07 KB
/
README.Rmd
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
---
output: github_document
---
<!-- README.md is generated from README.Rmd. Please edit that file -->
```{r, include = FALSE}
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>",
fig.path = "man/figures/README-",
out.width = "100%"
)
```
# hitype <a href="https://pwwang.github.io/hitype/"><img src="man/figures/logo.png" align="right" height="139" alt="hitype website" /></a>
<!-- badges: start -->
<!-- badges: end -->
**Hi**erarchical and **hi**gh-resolution cell-type identification for single-cell RNA-seq data inspired by [ScType][1].
## Features
- [x] Compatibility with [ScType][1]
- [x] Hierarchical and high-resolution cell-type identification
- [x] Train weights for your markers with a reference dataset
- [x] Speed optimization
- [x] Support as an R package with unit tests
[1]: https://github.com/IanevskiAleksandr/sc-type
## Installation
You can install the development version of `hitype` like so:
``` r
if (!requireNamespace("devtools", quietly = TRUE)) {
install.packages("devtools")
}
devtools::install_github("pwwang/hitype")
```
## Quick start
### Prepare the dataset
See also <https://satijalab.org/seurat/articles/pbmc3k_tutorial.html#setup-the-seurat-object>
<details>
<summary>Click to expand</summary>
```{r, warning=FALSE, message=FALSE}
pbmc <- pbmc3k.SeuratData::pbmc3k
pbmc[["percent.mt"]] <- Seurat::PercentageFeatureSet(pbmc, pattern = "^MT-")
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
pbmc <- Seurat::NormalizeData(pbmc)
pbmc <- Seurat::FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
pbmc <- Seurat::ScaleData(pbmc, features = rownames(pbmc))
pbmc <- Seurat::RunPCA(pbmc, features = Seurat::VariableFeatures(object = pbmc))
pbmc <- Seurat::FindNeighbors(pbmc, dims = 1:10)
pbmc <- Seurat::FindClusters(pbmc, resolution = 0.5)
pbmc <- Seurat::RunUMAP(pbmc, dims = 1:10)
```
</details>
### Use as a Seurat extension
```{r, warning=FALSE, out.width="75%"}
library(hitype)
# Load gene sets
gs <- gs_prepare(hitypedb_pbmc3k)
# Assign cell types
obj <- RunHitype(pbmc, gs)
Seurat::DimPlot(obj, group.by = "hitype", label = TRUE, label.box = TRUE) +
Seurat::NoLegend()
```
Compared to the manual marked cell types:
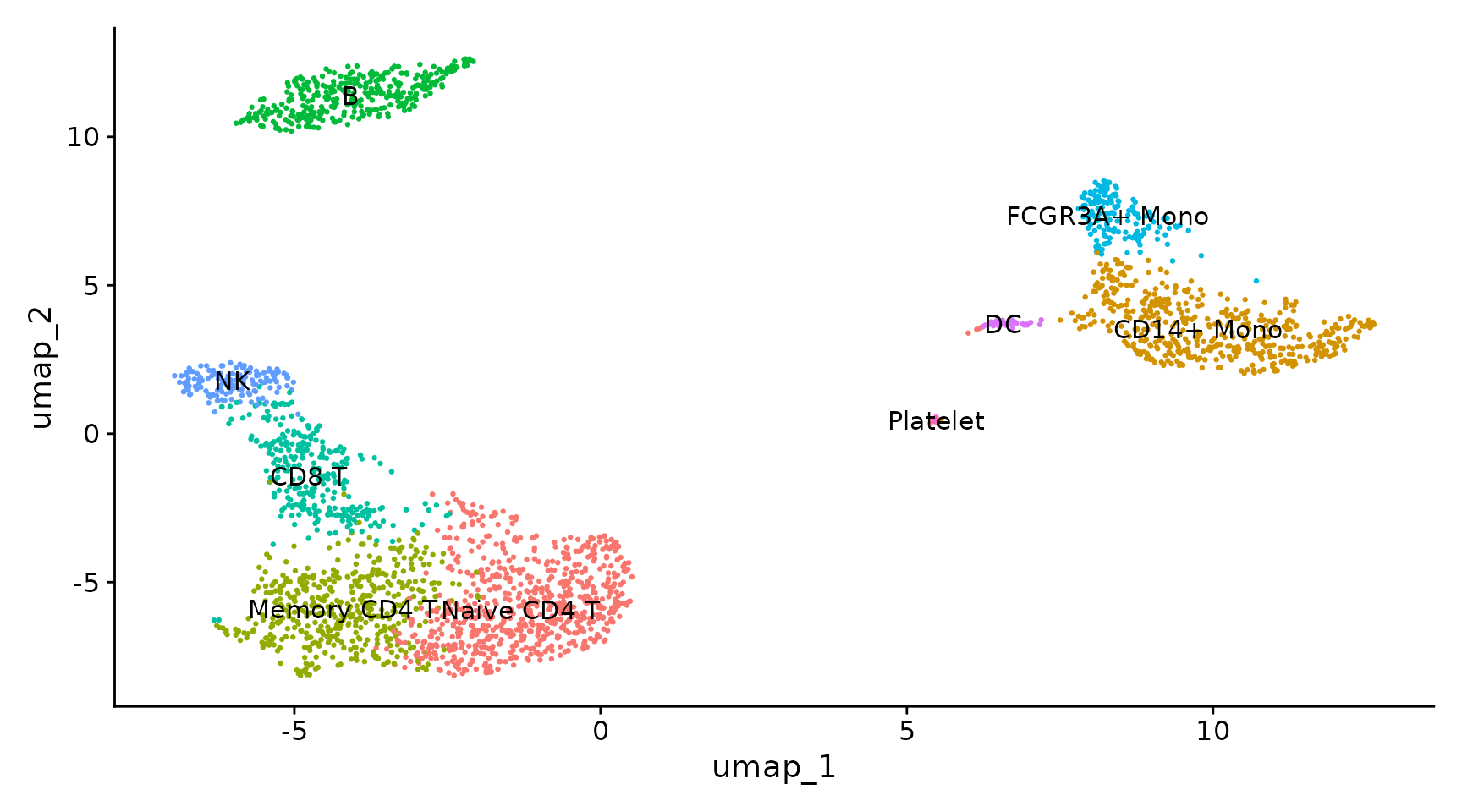{width=75%}
See also <https://satijalab.org/seurat/articles/pbmc3k_tutorial.html#assigning-cell-type-identity-to-clusters>
### Use as standalone functions
```{r, warning=FALSE}
scores <- hitype_score(pbmc@assays$RNA@scale.data, gs, scaled = TRUE)
cell_types <- hitype_assign(pbmc$seurat_clusters, scores, gs)
summary(cell_types)
```
You may see that we have exactly the same assignment in the Seurat tutorial:
|Cluster ID | Markers | Cell Type |
|:----------|:--------|:----------|
|0| IL7R, CCR7 | Naive CD4+ T |
|1| CD14, LYZ | CD14+ Mono|
|2| IL7R, S100A4 | Memory CD4+|
|3| MS4A1 | B|
|4| CD8A | CD8+ T|
|5| FCGR3A, MS4A7 | FCGR3A+ Mono|
|6| GNLY, NKG7 | NK|
|7| FCER1A, CST3 | DC|
|8| PPBP | Platelet|
## Documentation
<https://pwwang.github.io/hitype/>