| date | comments |
|---|---|
2024-11-10 |
true |
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。

本周话题: 7年两次考核未过,高校副教授亲历“非升即走”
这篇文章像是一部学术界的“生存游戏”纪实,主角马梅副教授经历了从满怀希望到失望离场的跌宕起伏。她如同科研战场的勇士,手握百万经费、发表顶刊论文,却最终在“非升即走”的关卡前败下阵来。文章揭示了学术圈既光鲜又残酷的一面,让人不禁感慨:在追求学术卓越的路上,有时不仅需要才华和努力,更需要一点运气和策略。马梅副教授的选择,则像是对传统路径的一次“叛逃”,她以新的身份重新开始,追寻内心的热爱与理想,让人看到了一个学者不屈不挠、勇于探索的精神风貌。
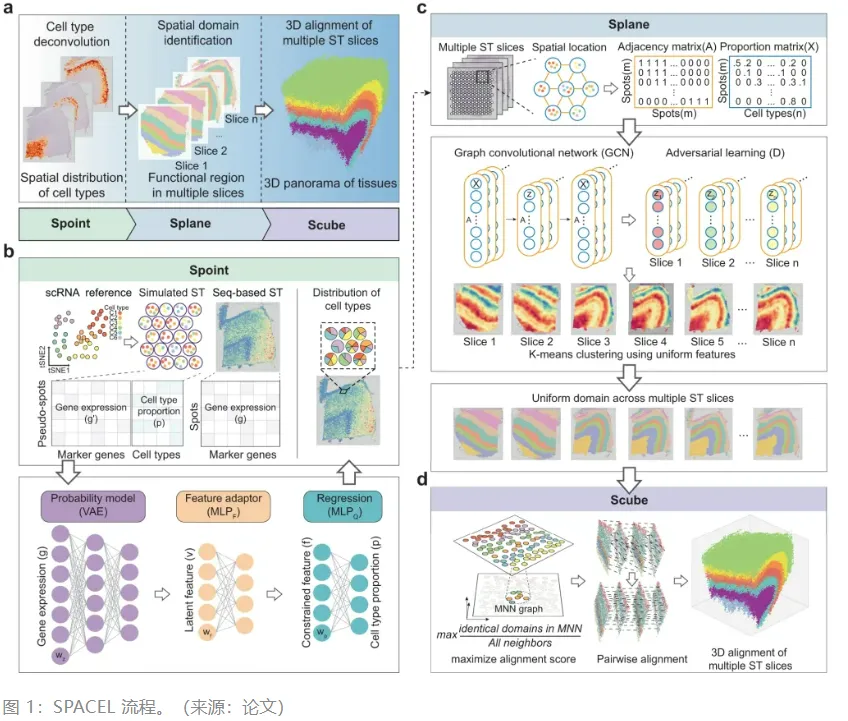
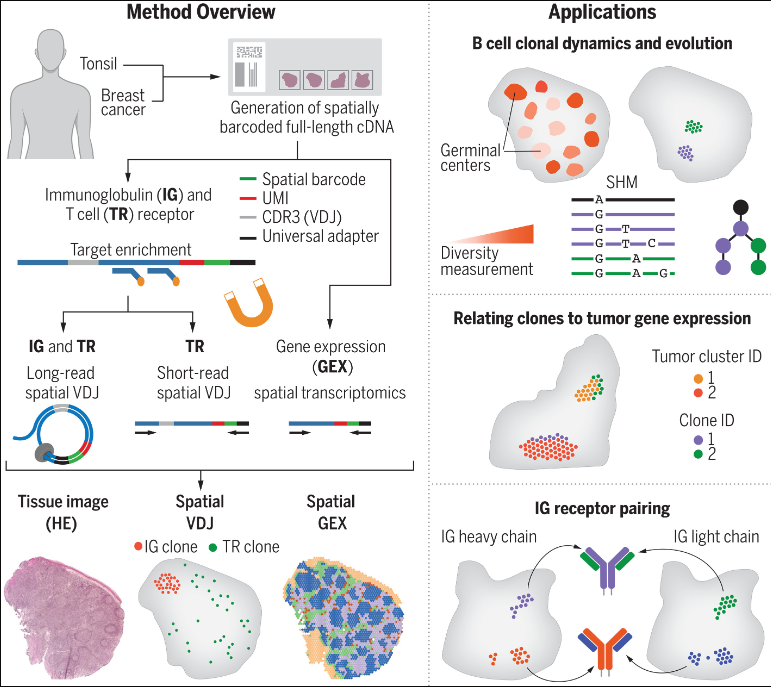
文章在Visium空间转录学(Spatial GEX)的基础上开发了VDJ序列空间转录组学(Spatial VDJ),它可同时捕获人类组织中的B细胞和T细胞抗原受体序列、并揭示细胞克隆动态,为研究感染、疫苗接种以及癌症的免疫反应提供新的工具。
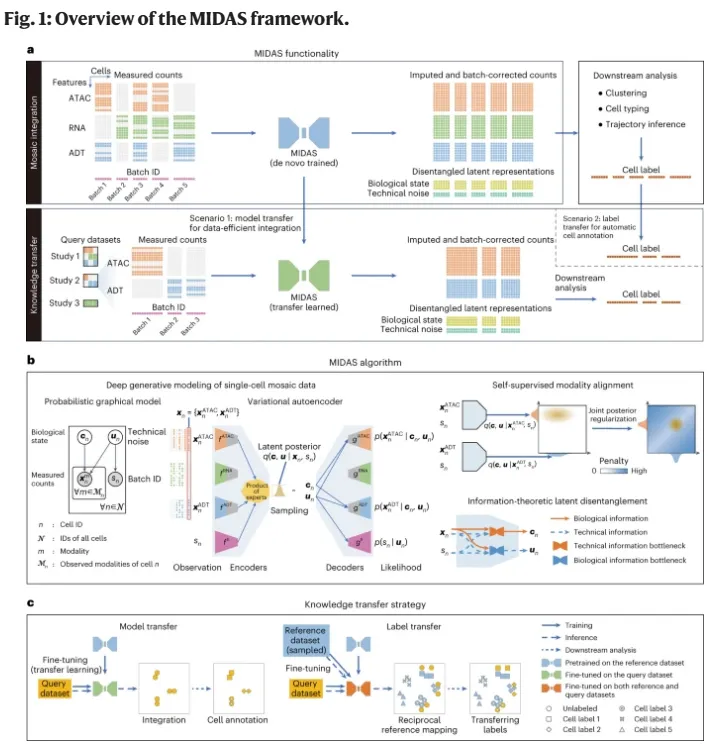
随着测序技术的发展和测序数据的增长,不同组学组合、不同测序技术、不同测序样本的“马赛克”式单细胞数据的整合成为巨大的挑战。为此,应晓敏团队和伯晓晨团队自主研发了基于生成式人工智能的新算法MIDAS。MIDAS假设每个细胞的多模态观测值是通过深度神经网络从两个与模态无关且解耦的隐变量生成的(即代表细胞异质性的生物状态,以及由单细胞实验引起的技术噪声),其输入由不同单细胞样本(批次)的表达矩阵和批次编号向量组成。这些批次可能来自不同的实验,或是不同的测序技术(例如CITE-seq和ASAP-seq),因此可能存在不同的技术噪声、模态组合和观测特征。MIDAS的输出包括生物状态和技术噪声两种低维表示的矩阵,以及对缺失模态和特征进行了补全并消除了批次效应的表达矩阵。这些输出可以用于聚类、细胞分型、轨迹推断等下游分析。
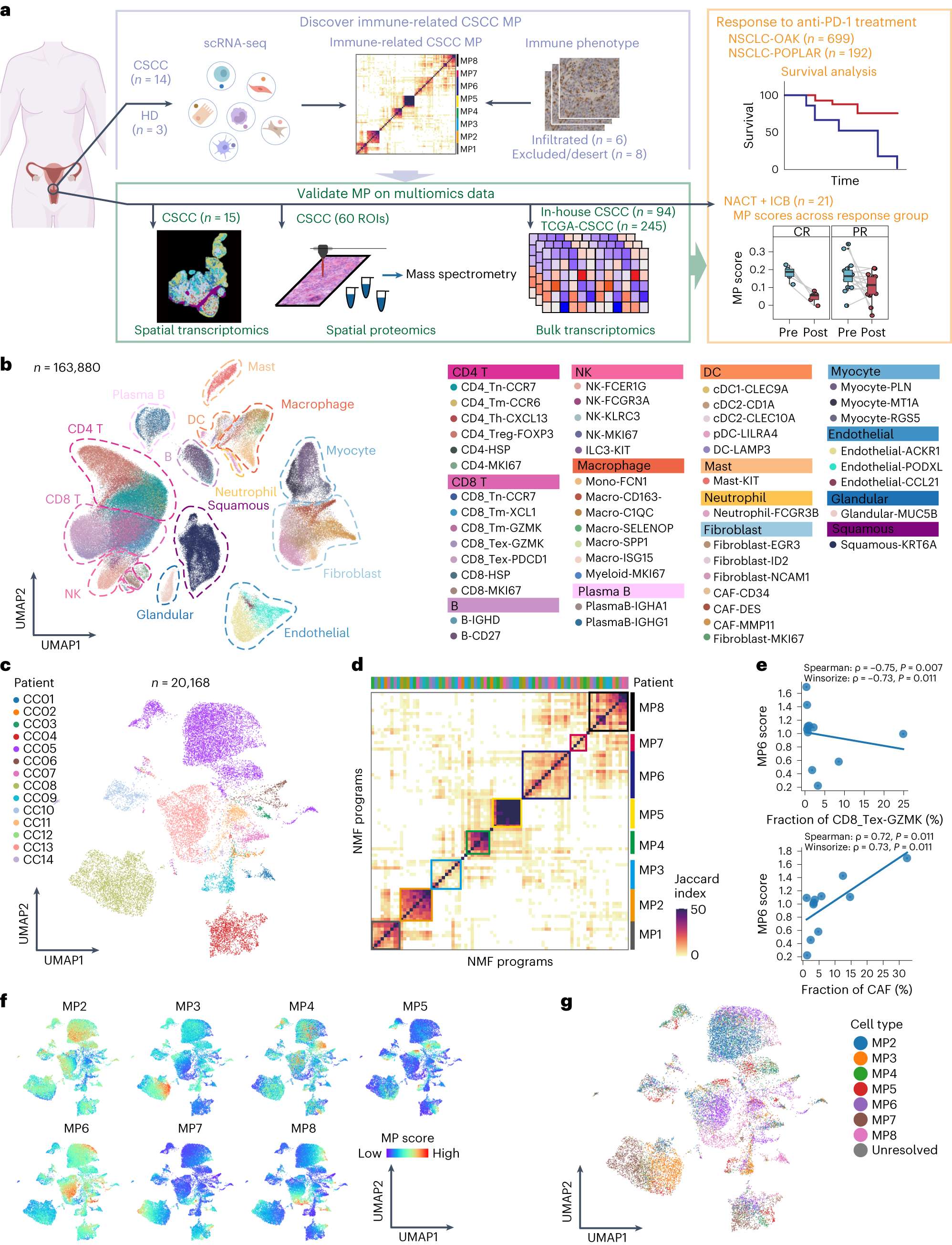
文章使用单细胞RNA测序技术对来自14个未经治疗的宫颈鳞状细胞癌样本的近163900个单细胞进行了测序,并利用空间转录组与空间蛋白组学绘制了宫颈鳞状细胞癌内表达异质性的高分辨率和空间分辨图谱。

近日,胃肠道健康诊断检测开发公司Geneoscopy宣布美国FDA批准了其非侵入性结直肠癌(CRC)筛查检测ColoSense。ColoSense适用于45岁或以上具有CRC典型平均风险的成年人。ColoSense被FDA指定为突破性设备,是首个利用RNA生物标志物提供疾病活动动态视图的无创性CRC筛查检测。
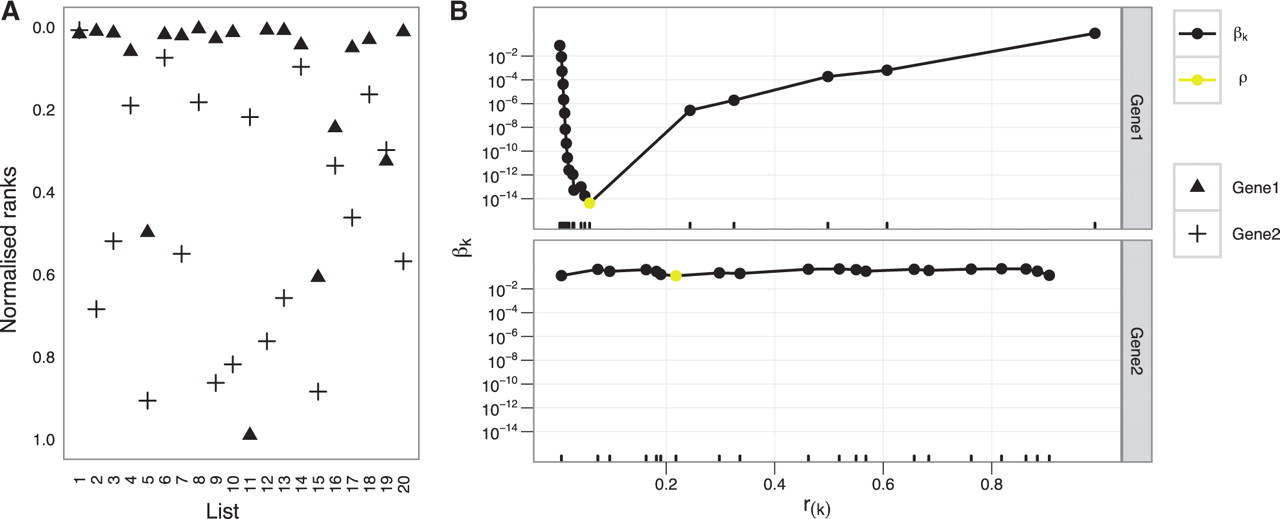
RRA(Robust Rank Aggregation)是一种对排名进行整合,获得一个综合性排名列表的算法。文中使用具体例子展现了RRA的实现过程。

2023年9月23日,BioLinkX于线上腾讯会议举办了生信半月谈活动。本次活动的主题是肿瘤新抗原及其在临床治疗中的应用,主讲人为南京澄实生物生物信息副总监,吴增丁老师。 活动中主讲人介绍了肿瘤新抗原的概念及其临床背景,详细介绍了新抗原的来源和检测方法,展示了NetMHC的应用,最后概述了新抗原的验证方法。
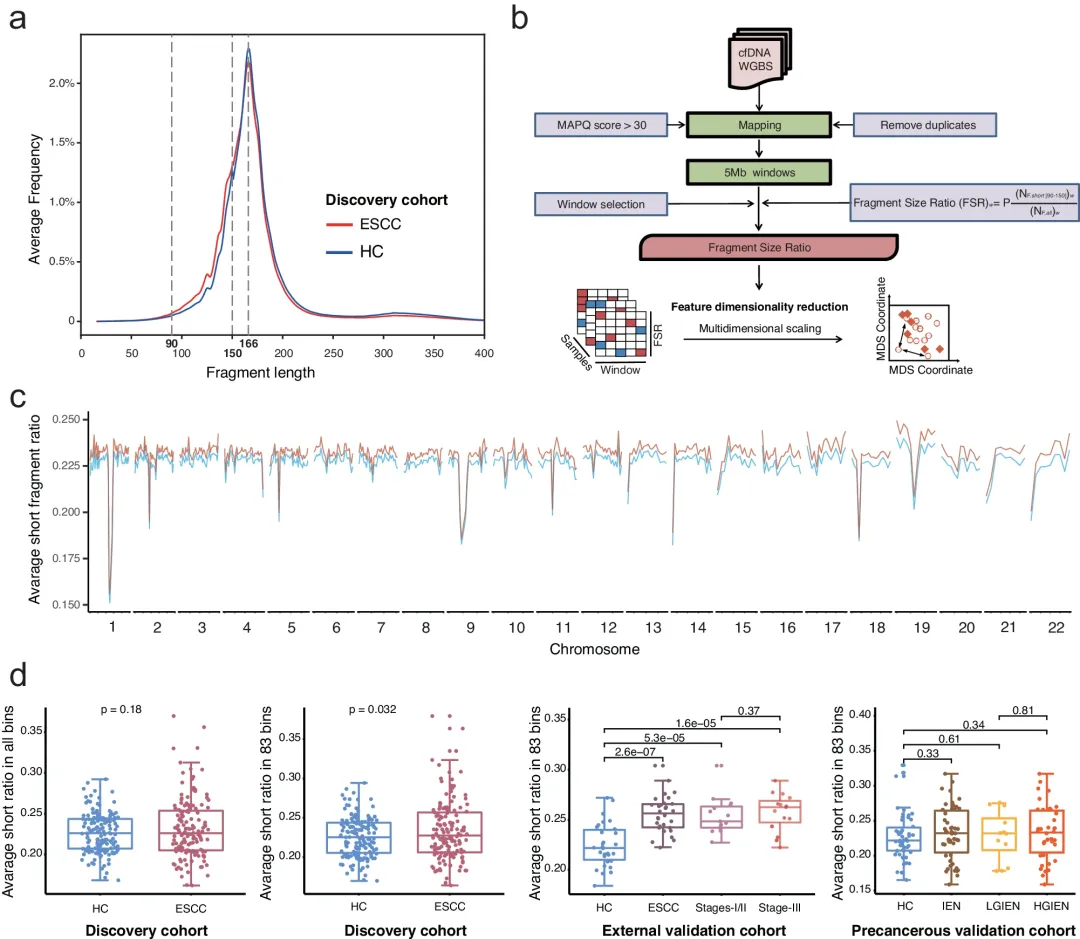
食管鳞状细胞癌(ESCC)是食管癌(EC)的主要组织学亚型,约占新发EC病例的88%,具有异质性高、进展快、预后差等特点;早期ESCC患者缺乏特异症状,首次就诊时多处于中晚期,5年生存率较低。 中国医学科学院肿瘤医院刘芝华、陈洪岩团队开发了一种名为EMMA的综合分析方法,能同时识别差异甲基化区域(DMRs)、CNVs和片段化特征,以实现ESCC的超早期检测。该方法使人们能够分析cfDNA中ESCC的表观遗传和遗传特征的互补性、时间动态和检测效率,可确定最佳cfDNA甲基化特征的生物学相关性。该模型不仅显著提高了ESCC的无创检测能力,还具有动态分子监测和指导治疗潜在价值。
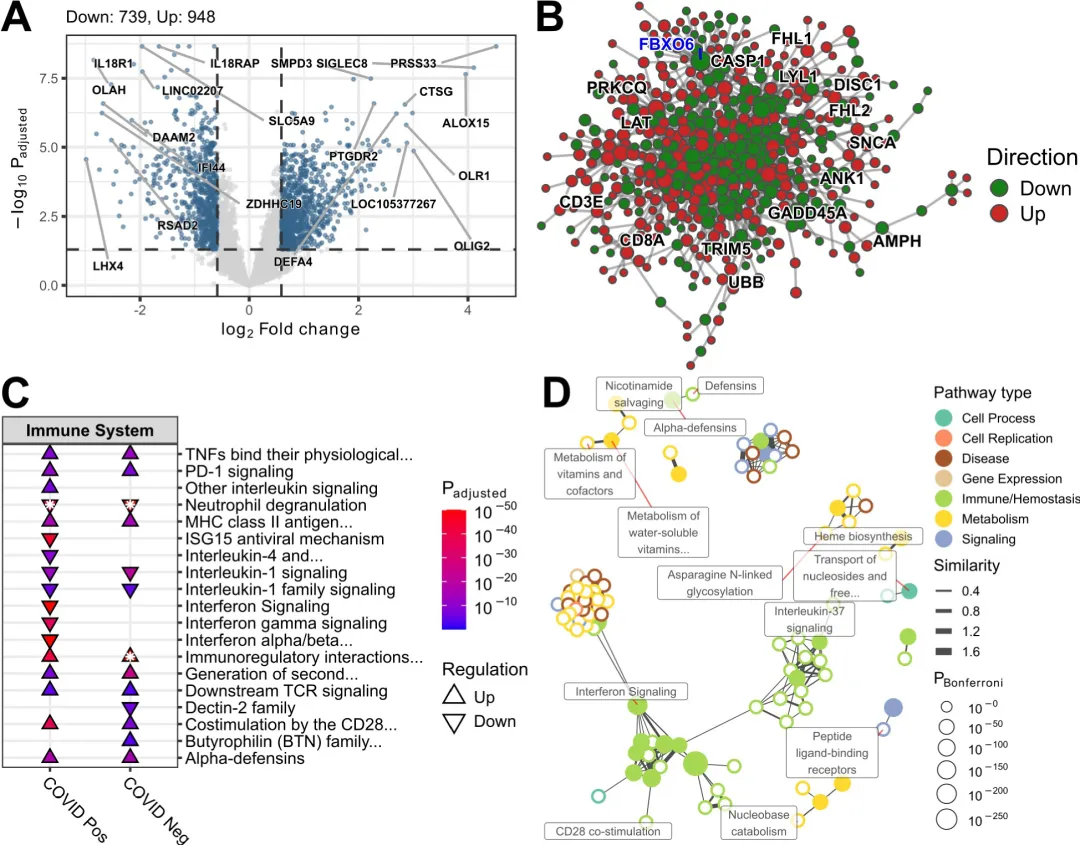
仅凭差异表达基因(differentially expressed genes,DEG)的列表本身不足以得出有关其作用机制的有意义结论,必须对其进行进一步分析。有几种路径可供选择,其中最常见的两种方法是通路富集和网络分析,这些可以结合起来进一步提取生物学意义。Blimkie等人介绍了一个R包pathlinkR,它提供了一个简化和统一的界面来执行一些最常见的DEG分析和可视化,其功能可以帮助用户轻松执行通路富集和网络分析。它被设计成易于使用,并接受典型分析管道中最常用工具DESeq2和edgeR的输入。DESeq2和edgeR两个软件包都会生成一个DEG表,pathlinkR无需修改即可使用该表输入进行核心功能分析,包括可视化、通路富集和蛋白质-蛋白质相互作用(PPI)网络的构建。
- 工具:https://bioconductor.org/packages/pathlinkR/
- https://github.com/hancockinformatics/pathlinkR/
- 文献:Blimkie TM, An A, Hancock REW. Facilitating pathway and network based analysis of RNA-Seq data with pathlinkR. PLoS Comput Biol. 2024 Sep 16;20(9):e1012422. doi: 10.1371/journal.pcbi.1012422.

AGAT 能够检查、修复、填充任何类型的 GTF 和 GFF 的缺失信息(特征/属性),以创建完整、排序和标准化的 gff3 格式。多年来,它已经被许多工具丰富起来,几乎可以执行与 GTF/GFF 格式文件相关的任何可能的任务(清理、转换、合并、修改、过滤、FASTA 序列提取、添加信息等)。与其他方法相比,AGAT 甚至可以对最复杂的 GTF/GFF 文件保持最好的效果。
文档:https://nbisweden.github.io/AGAT/
「Openbiox 生信周刊」运维小队:
- @ShixiangWang(王诗翔)
- @kkjtmac(阚科佳)
- @NiEntropy(赵启祥)
- @He-Kai-fly(何凯)
- @JnanZhang(张佳楠)
- @kkjtmac(阚科佳)
- @Tomcxf(陈啸枫)
- @wangdepin(王德品)
- @kongjianyang(空间阳)
- @donghongyu2020(董弘禹)
- @DrRobinLuo(罗鹏)
- @Wangcy-rachel - 王春阳
- @zoe3251 - 舒晨阳
这个周刊每周日发布,同步更新在微信公众号「优雅R」(elegant-r)上。
微信搜索“优雅R”或者扫描二维码,即可订阅。
